Linfoma de Hodgkin vs no Hodgkin
 El linfoma de Hodgkin
(LH) comprende un grupo de neoplasias linfoides que difieren del LNH en varios
aspectos. Si bien el LNH es frecuente en localizaciones extraganglionares y se disemina siguiendo
un patrón impredecible, el LH surge en un único ganglio o cadena ganglionar y se disemina primero
hacia los tejidos linfoides contiguos
anatómicamente. Por
este motivo, la estadificación del LH es mucho más importante para orientar el
tratamiento que en el caso del LNH. El LH también tiene unas características
morfológicas distintivas. Se caracteriza por la presencia de unas células
gigantes neoplásicas denominadas células de Reed-Sternberg. Esas células liberan factores que
inducen la acumulación de linfocitos, macrófagos y granulocitos reactivos que
suponen más del 90% de la celularidad tumoral. En la inmensa mayoría de los LH,
las células neoplásicas de Reed-Sternberg derivan linfocitos B del centro
germinal o poscentro germinal.
El linfoma de Hodgkin
(LH) comprende un grupo de neoplasias linfoides que difieren del LNH en varios
aspectos. Si bien el LNH es frecuente en localizaciones extraganglionares y se disemina siguiendo
un patrón impredecible, el LH surge en un único ganglio o cadena ganglionar y se disemina primero
hacia los tejidos linfoides contiguos
anatómicamente. Por
este motivo, la estadificación del LH es mucho más importante para orientar el
tratamiento que en el caso del LNH. El LH también tiene unas características
morfológicas distintivas. Se caracteriza por la presencia de unas células
gigantes neoplásicas denominadas células de Reed-Sternberg. Esas células liberan factores que
inducen la acumulación de linfocitos, macrófagos y granulocitos reactivos que
suponen más del 90% de la celularidad tumoral. En la inmensa mayoría de los LH,
las células neoplásicas de Reed-Sternberg derivan linfocitos B del centro
germinal o poscentro germinal.
El linfoma de Hodgkin
es responsable del 0,7% de todos los cánceres nuevos en EE. UU., con 8.000
casos nuevos cada año. La edad media en el momento del diagnóstico es de 32
años. Es uno de los cánceres más frecuentes de los adultos jóvenes y
adolescentes, pero también aparece en ancianos. Fue el primer cáncer en el
hombre que fue tratado con éxito con radioterapia y quimioterapia, y es curable
en la mayoría de los casos.
Clasificación . En la clasificación
de la OMS se reconocen cinco subtipos de LH:
1. Esclerosis nodular
2. Celularidad mixta
3. Rico en linfocitos
4. Con depleción
linfocítica
5. De predominio
linfocítico
En los primeros
cuatro subtipos, esclerosis nodular, celularidad mixta, rico en linfocitos y
con depleción linfocítica, las células de Reed-Sternberg tienen un inmunofenotipo
similar. Esos subtipos se agrupan como formas clásicas de LH. En el subtipo restante, de
predominio linfocítico, las células de Reed-Sternberg tienen un inmunofenotipo
diferenciado de linfocitos B que difiere del encontrado en los tipos
«clásicos».
Morfologia.
La identificación de
células de Reed-Sternberg y sus variantes es esencial para el diagnóstico. Las
celulas diagnosticas de
Reed-Sternberg son celulas grandes ( ≥ 45 mm de diametro) con multiples nucleos o un
solo nucleo con multiples lobulos nucleares, cada uno con un gran
nucleolo a modo
de
inclusion con el tamano aproximado de un linfocito pequeño
(5-7 mm
de diametro).
El citoplasma es abundante. Asimismo, se reconocen algunas variantes de las
células de Reed-Sternberg. Las variantes mononucleares contienen un único
núcleo con un gran nucléolo a modo de inclusión. Las células lacunares (que se ven en el
subtipo de esclerosis nodular) tienen núcleos más delicados, plegados o
multilobulares y un citoplasma pálido abundante que se ve a menudo alterado
durante el corte de los cortes, dejando el núcleo asentado en un agujero vacío
(una laguna).
En las formas clásicas de LH, las células de
Reed-Sternberg sufren una forma peculiar de muerte célula en la cual las
células pierden volumen y se tornan picnóticas, un proceso que se describe como
la «momificación». Las variantes linfohistocíticas (células L-H) con núcleos
polipoides, nucléolos poco notorios y un citoplasma moderadamente abundante son
características del subtipo de predominio linfocítico.
El LH debe distinguirse de otras afecciones
en las que se pueden ver células parecidas a las células de Reed-Sternberg, como
la mononucleosis infecciosa, cánceres de tejidos sólidos y el LNH de células
grandes. El diagnóstico de LH depende de la identificación de las células de
Reed-Sternberg en un fondo típico prominente de células inflamatorias no neoplásicas.
Las células de Reed-Sternberg del LH también tienen un perfil
inmunohistoquímico característico.

Tipo de esclerosis
nodular. Es
la forma más frecuente de LH, que supone el 65-70% de los casos. Se caracteriza
por la presencia de células de Reed-Sternberg de la variante
lacunar y
por el deposito de colageno en bandas que
dividen los ganglios
linfaticos afectados en nodulos circunscritos. La fibrosis puede ser escasa o abundante.
Las células de Reed-Sternberg se encuentran sobre un fondo polimorfo de
linfocitos T, eosinófilos, células plasmáticas y macrófagos. Es frecuente
encontrar células diagnósticas de Reed-Sternberg. Las células de Reed-Sternberg
en este y otros subtipos «clásicos» del LH poseen un inmunofenotipo característico.
Son positivas para los factores PAX5 (un factor de transcripción de los
linfocitos B), CD15 y CD30, y negativas para otros marcadores de los linfocitos
B y de los linfocitos T y CD45 (antígeno leucocitario común). Al igual que en
otras formas de LH, la afectación del bazo, hígado, médula ósea y otros órganos
y tejidos puede aparecer a su debido tiempo en forma de nódulos tumorales
irregulares que se parecen a los que se ven en los
ganglios linfáticos. Este subtipo se asocia en raras ocasiones al VEB.
El
tipo de esclerosis nodular se presenta con igual frecuencia en hombres y mujeres,
con una cierta tendencia a afectar los ganglios linfáticos cervicales bajos,
supraclaviculares y mediastínicos de adolescentes o adultos jóvenes. El
pronóstico es excelente.
Tipo de celularidad mixta. Esta
forma de LH constituye el 20-25% de los casos. Los ganglios linfáticos
afectados se ven borrados difusamente por un infiltrado celular heterogéneo que
contiene linfocitos T, eosinófilos, células plasmáticas y macrófagos benignos, mezclados
con células de Reed-Sternberg. Las celulas diagnosticas de
Reed-Sternberg y las variantes mononucleares son abundantes. Las celulas de
Reed-Sternberg estan infectadas por el VEB en el 70% de los casos. El
inmunofenotipo es
idéntico al observado en el tipo de esclerosis nodular. El
LH de celularidad mixta es más frecuente en los varones.
Comparado
con los subtipos de predominio linfocítico y esclerosis nodular, es más
probable que se asocie a una mayor edad, a síntomas sistémicos, como sudoración
nocturna y pérdida de peso, y a un estadio tumoral avanzado. No obstante, el
pronóstico global es muy bueno.
Tipo rico en linfocitos. Se
trata de una forma infrecuente del LH clásico en el que los linfocitos
reactivos suponen la inmensa mayoría del infiltrado celular. En la mayor parte
de los casos, los ganglios linfáticos afectados están borrados difusamente,
pero con una nodularidad vaga, porque a veces pueden verse folículos residuales
de linfocitos B. Esta entidad se distingue del tipo de predominio linfocítico
por la presencia de variantes mononucleares frecuentes y de células diagnósticas
de Reed-Sternberg con un perfil inmunofenotípico «clásico».
Se asocia al VEB en el 40% de los casos, y su pronóstico es muy bueno o
excelente.
Tipo con depleción linfocítica. Se
trata del tipo menos frecuente de LH, suponiendo menos del 5% de los casos. Se
caracteriza por la escasez de linfocitos y la abundancia relativa de células de
Reed-Sternberg o de sus variantes pleomorfas. El inmunofenotipo de las células
de Reed-Sternberg es idéntico al observadoen otros tipos clásicos de LH. El
inmunofenotipado es esencial, ya que la mayoría de los tumores sospechosos de
pertenecer al tipo de depleción linfocítica demuestra ser en realidad un LNH de
células grandes. Las células de Reed-Sternberg están infectadas por el VEB en
más del 90% de los casos.
El
LH con depleción linfocítica se presenta predominantemente en los ancianos, en
sujetos VIH+ de cualquier edad y en países no industrializados. Es frecuente
encontrar casos en estadio avanzado y con síntomas sistémicos, y la evolución general
es algo menos favorable que en los demás subtipos.
Tipo de predominio linfocítico. Esta
variante «no clásica» frecuente de LH es responsable del 5% de los casos. Los ganglios
afectados están difuminados por un infiltrado nodular de linfocitos pequeños
mezclados con un número variable de macrófagos. Las células de Reed-Sternberg «clásicas»
son normalmente difíciles de encontrar, por el contrario, este tumor contiene
las variantes denominadas L-H (linfocítica e histiocítica), que tienen un
núcleo multilobulado que se parece a una palomita de maíz («célula en palomita
de maíz»). Los eosinófilos y células plasmáticas son normalmente escasos o
ausentes.
Al
contrario que las células de Reed-Sternberg que se encuentran en las formas
clásicas del LH, las
variantes L-H expresan marcadores tipicos de los linfocitos B del centro germinal,
como CD20 y BCL6, y son normalmente negativas para CD15 y CD30. El patrón
nodular de crecimiento se debe a la presencia de folículos expandidos de linfocitos
B, que están llenos de variantes L-H, y numerosos linfocitos B y células dendríticas
foliculares reactivas. Los genes IgH de las variantes L-H muestran indicios de
hipermutación somática continuada, una modificación que se presenta sólo en los
linfocitos B del centro germinal. En el 3-5% de los casos, este tipo se transforma
en un tumor que se parece al linfoma difuso de linfocitos B grandes. El VEB no
se asocia a este subtipo.
La
mayoría de los pacientes son varones, normalmente menores de 35 años de edad,
que acuden típicamente con linfadenopatías cervicales o axilares. La afectación
del mediastino y la médula ósea es rara. En algunas series, las probabilidadesde
recurrencia de esta forma de LH son mayores que las de los subtipos clásicos,
pero el pronóstico es excelente.
Patogenia
molecular . El origen de las células neoplásicas de
Reed-Sternberg del LH clásico se han explicado mediante elegantes estudios que
se basan en el análisis molecular de las células de Reed-Sternberg y variantes
aisladas. En la inmensa mayoría de los casos, los genes Ig
de las células de Reed-Sternberg han sufrido tanto la recombinación V(D)J como la hipermutación somática que
establecen su origen desde un
linfocito B del centro germinal o poscentro germinal.A pesar de tener el mapa genético de un linfocito B, las
células Reed-Sternberg del LH clásico no expresan la mayoría de los genes específicos
del linfocito B, incluidos los genes de Ig. Se desconoce la causa de esta
reprogramación masiva de la expresión génica. La
activación del factor de transcripción NF- k B es un episodio habitual en el LH clásico. El NF- k B se
activa por la infección por el VEB o por algún otro mecanismo, y activa genes
que favorecen la supervivencia y proliferación del linfocito. Las células
tumorales VEB+ expresan la proteína 1 latente de membrana (LMP-1), una
proteína codificada por el genoma del VEB que transmite señales que estimulan
el NF- k B. A su vez, la activación del NF- k B también tiene lugar en tumores VEB −
, en algunos casos como consecuencia del resultado de
mutaciones adquiridas en el I k B,
un regulador negativo del NF-kê B. Se ha propuesto que la activación del NF- k B por el VEB u otros mecanismos permite el rescate de los
linfocitos B del centro germinal «tullidos» que no pueden expresar las Ig por apoptosis,
creando el escenario para la adquisición de otras mutaciones desconocidas que
colaboran para producir las células de Reed-Sternberg. Sabemos poco sobre las
bases de la morfología de las células de Reed-Sternberg y sus variantes, pero
es interesante encontrar linfocitos B infectados por el VEB parecidos a las
células de Reed-Sternberg en los ganglios linfáticos de sujetos con
mononucleosis infecciosa, lo que indicaría que las proteínas codificadas por el
VEB participan en la excepcional metamorfosis de los linfocitos B en células de
Reed-Sternberg.
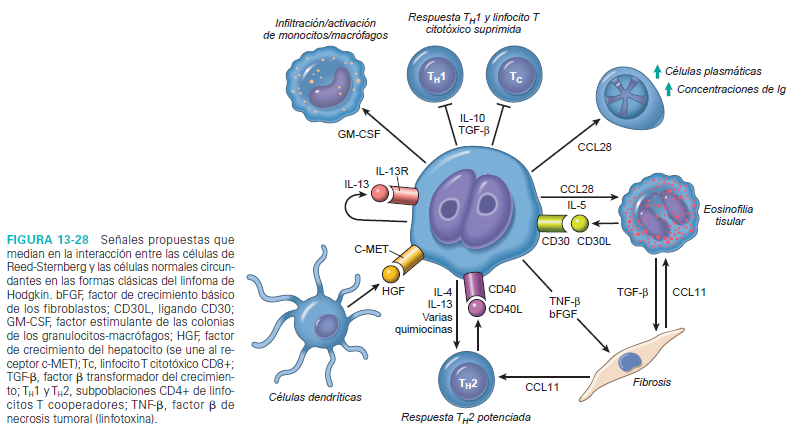
La acumulación florida de las células reactivas en los
tejidos afectados por el LH clásico se produce en respuesta a una amplia variedad
de citocinas (como IL-5, IL-10,
IL-13 y TGF- beta) y quimiocinas (como TARC, MDC, IP10 y CCL28) segregadas por las células
de Reed-Sternberg. Una vez
atraídas, las células reactivas producen factores que apoyan el crecimiento y
la supervivencia de las células tumorales y modifican aún más la respuesta de
la célula reactiva. Por ejemplo, los eosinófilos y linfocitos T expresan ligandos
que activan los receptores CD30 y CD40 de las células de Reed-Sternberg,
produciendo señales que estimulan el NF- k B. Las células de
Reed-Sternberg son aneuploides y poseen diversas aberraciones cromosómicas
clonales. Es particularmente frecuente encontrar un mayor número de copias
acumuladas en el protooncogén c-REL en el cromosoma 2p, lo que podría contribuir al aumento de
la actividad NF- k B.
Características
clínicas . El LH se presenta con mayor frecuencia como
una linfadenopatía indolora. Los pacientes con los tipos de esclerosis nodular
o predominio linfocítico se presentan con enfermedad en estadio I-II,
normalmente sin manifestaciones sistémicas.
Los pacientes con enfermedad diseminada (estadios III-IV) o
con los subtipos de celularidad mixta o depleción linfocítica se presentan más
a menudo con síntomas constitucionales, como fiebre, sudores nocturnos y
pérdida de peso. En la mayoría de los casos puede encontrarse anergia cutánea
como consecuencia de la depresión de la inmunidad celular. La mezcla de
factores liberados de las células de Reed-Sternberg suprime
la respuesta inmunitaria T H 1 y contribuye a la alteración inmunitaria.
La diseminación del LH sigue un estereotipo notable: primero
la enfermedad ganglionar, luego la enfermedad esplénica, la enfermedad hepática
y, por último, la afectación de la médula y otros tejidos.
Debido a su comportamiento, la radioterapia puede ser
curativa en los estadios iniciales, por lo cual la estadificación del LH no sólo
determina el pronóstico, sino que también dirige el tratamiento.
La estadificación consiste en exploración física, estudio radiológico
del abdomen, pelvis y tórax, y biopsia de médula ósea. Siempre que la
estadificación sea dudosa, se prefiere el tratamiento sistémico.
Con los protocolos actuales de tratamiento, la variable
pronóstica más importante es el estadio tumoral más que el tipo histológico. La
tasa de curación en los estadios I y IIA se acerca al 90%. Incluso en la
enfermedad avanzada (estadios IVA y IVB), la supervivencia sin enfermedad a 5
años es del 60-70%.
Los avances introducidos en el tratamiento del LH han dado
paso a nuevos problemas. Los supervivientes a largo plazo de la quimioterapia y
la radioterapia tienen un mayor riesgo de desarrollar segundos cánceres. Los
síndromes mielodisplásicos, la LMA y el cáncer de pulmón encabezan la lista,
pero también se producen LNH, cáncer de mama, cáncer gástrico, sarcomas y
melanomas. La mayor parte del riesgo de los tumores sólidos se atribuye a la
radioterapia, que también se ha relacionado con la fibrosis pulmonar y con la
aceleración de la aterosclerosis. El riesgo de cáncer de mama es
particularmente alto en las mujeres tratadas con radioterapia en el tórax
durante la adolescencia. Los fármacos alquilantes parecen ser responsables del
aumento de riesgo de LMA y mielodisplasia. Por fortuna, las combinaciones más
modernas de fármacos quimioterápicos y el empleo más prudente de la
radioterapia parecen evitar gran parte de esas complicaciones, y son
igualmente curativas.
BIBLIOGRAFIA: "Patologia estructural y funcional", Robbins y Cotran, 8va edicion.


